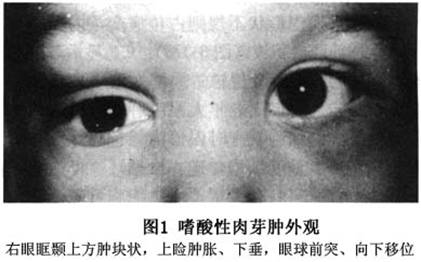
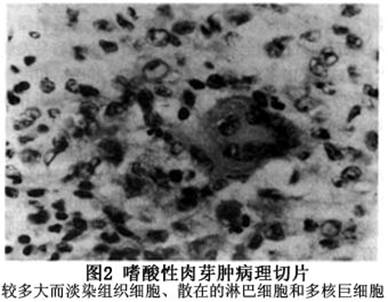
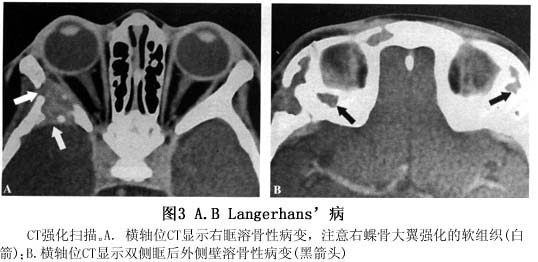
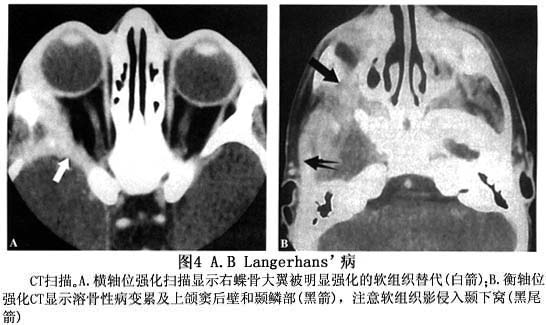
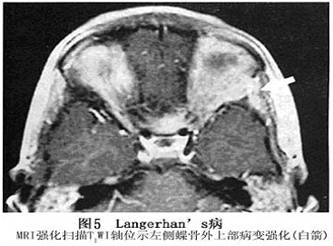
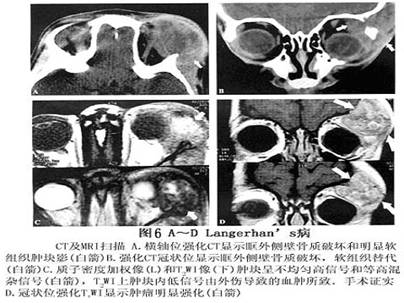
眼眶嗜酸性肉芽肿
概述:组织细胞增多病X(
histiocytosis X)是由大量增生的染色淡而大的组织细胞在眼眶或全身各脏器、软组织和骨中堆积引起的病变。根据累及的部位不同又分为局部单灶性和全身多灶性。年龄越小越容易发展成为多灶性病灶,患者年龄偏大,单灶性病变可能性大。1~2岁内多系统如皮肤、内脏、淋巴结和眼眶受侵犯者视为
累-赛病(Letterer-Siwe disease);年龄稍大的小儿表现为典型的三联征:
颅骨缺损,突眼和
尿崩症称为
汉德-许勒尔-克思斯琴病(Hand-Schüller-Christian disease);单一性病变叫嗜酸性细胞肉芽肿(
eosinophilic granuloma)。众多学者对该病进行了深入研究,从临床表现和大量的超微结构和免疫组织化学的资料来看,现有倾向用朗格汉斯细胞组织细胞增多病(Langerhans cell histiocytosis)代替组织细胞增多病X。以前习惯性称以上3种为不同类型的病变,现认为Letterer-Siwe病和Hand-Schüller-Christian病只是相同疾病的不同过程,嗜酸性细胞肉芽肿可能代表一种反应型,认为该组病变类似于肉芽肿的良性过程,而不具有新生物的特性。根据预后好的特点,有理由将嗜酸性肉芽肿与其他两种分离出来。现未发现孤立性的嗜酸性肉芽肿发展成为系统性多灶性病变或因此病而死亡的患者。病变没有显示家族性,属于非遗传性疾病。急性或亚急性的严重病变可
自发消退,小剂量的皮质类固醇、放射治疗和抗肿瘤的化学治疗能抑制病变的进程。根据临床资料和病变的预后将该组织病变分为5类:
1.骨病变 50%的患者主要引起骨病变,单个或多个病灶倾向于累及轴性骨(头颅、脊柱、肩胛、骨盆和四肢近端长骨),多为嗜酸性细胞肉芽肿,预后良好。
2.骨病变和少量的全身病变 骨髓受累可导致贫血,基底脑膜病变可引起
尿崩症,黏膜和皮肤也受侵犯,此临床表现在Hand-Schüller-Christian病中多见。
3.骨病变加上中到重度的内脏损害 骨损害主要发生在颅骨的基底部、眶周骨和中耳骨;内脏病变包括肺、脾、肝以及淋巴结、黏膜和头颅皮肤受累,该病变在严重的Hand-Schüller-Christian病和轻度的Leterer-Siwe病患者中发生。
4.严重的内脏病变 肝、脾脏迅速而进行性肿大,肺脏病、贫血、血小板减少和皮疹也很快出现,因为髓内组织细胞快速增生,故骨破坏比慢性病例发生少,这些发现与Letterer-Siwe病相符合,预后差。
5.局限病变 这一小组患者一般很年轻,侵袭性病变发生在特殊的部位并伴有轻微的全身病变。这些病变可能是颈部的淋巴结病,肝脏病或少见的严重肺囊肿。


 流行病学
流行病学